Variant calling workflow
Overview
Teaching: 35 min
Exercises: 25 minLearning Objectives
Understand the steps involved in variant calling.
Describe the types of data formats encountered during variant calling.
Use command line tools to perform variant calling.
We mentioned before that we are working with files from a long-term evolution study of an E. coli population (designated Ara-3). Now that we have looked at our data to make sure that it is high quality, and removed low-quality base calls, we can perform variant calling to see how the population changed over time. We care how this population changed relative to the original population, E. coli strain REL606. Therefore, we will align each of our samples to the E. coli REL606 reference genome, and see what difference exist in our reads versus the genome.
Alignment to a reference genome
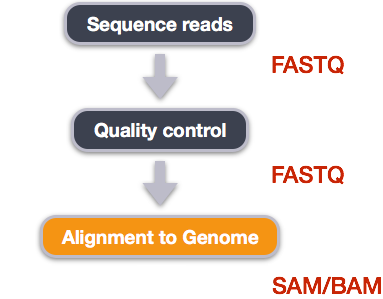
We perform read alignment or mapping to determine where in the genome our reads originated from. There are a number of tools to choose from and, while there is no gold standard, there are some tools that are better suited for particular NGS analyses. We will be using the bowtie2 algorithm for mapping these sequences against a reference genome.
The alignment process consists of two steps:
- Indexing the reference genome
- Aligning the reads to the reference genome
Setting up
First we download the reference genome for E. coli REL606. Although we could copy or move the file with cp or mv, most genomics workflows begin with a download step, so we will practice that here.
mkdir -p data/genome
curl -L -o data/genome/ecoli_rel606.fasta.gz ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/017/985/GCA_000017985.1_ASM1798v1/GCA_000017985.1_ASM1798v1_genomic.fna.gz
gunzip data/genome/ecoli_rel606.fasta.gz
Exercise
We saved this file as
data/genome/ecoli_rel606.fasta.gzand then decompressed it. What is the real name of the genome?Solution
$ head data/genome/ecoli_rel606.fastaThe name of the sequence follows the
>character. The name isCP000819.1 Escherichia coli B str. REL606, complete genome. Keep this chromosome name (CP000819.1) in mind, as we will use it later in the lesson.
We will also download a set of trimmed FASTQ files to work with. These are small subsets of our real trimmed data, and will enable us to run our variant calling workflow quite quickly.
# This is a place holder so the workflow will work. This will be updated with figshare links later
$ curl -L -o sub.tar.gz https://osf.io/gbt7p/download
$ tar xvf sub.tar.gz
$ mv sub/ ~/dc_workshop/data/trimmed_fastq_small
You will also need to create directories for the results that will be generated as part of this workflow. We can do this in a single
line of code because mkdir can accept multiple new directory
names as input.
mkdir -p results/sam results/bam results/bcf results/vcf
Index the reference genome
Our first step is to index the reference genome for use by BWA. Indexing allows the aligner to quickly find potential alignment sites for query sequences in a genome, which saves time during alignment. Indexing the reference only has to be run once. The only reason you would want to create a new index is if you are working with a different reference genome or you are using a different tool for alignment.
The index is built using bowtie2-build, whose options you can see by running it alone
bowtie2-build
## No input sequence or sequence file specified!
## Bowtie 2 version 2.3.4.1 by Ben Langmead (langmea@cs.jhu.edu, www.cs.jhu.edu/~langmea)
## Usage: bowtie2-build [options]* <reference_in> <bt2_index_base>
## reference_in comma-separated list of files with ref sequences
## bt2_index_base write bt2 data to files with this dir/basename
## *** Bowtie 2 indexes work only with v2 (not v1). Likewise for v1 indexes. ***
## Options:
## -f reference files are Fasta (default)
## -c reference sequences given on cmd line (as
## <reference_in>)
## --large-index force generated index to be 'large', even if ref
## has fewer than 4 billion nucleotides
## -a/--noauto disable automatic -p/--bmax/--dcv memory-fitting
## -p/--packed use packed strings internally; slower, less memory
## --bmax <int> max bucket sz for blockwise suffix-array builder
## --bmaxdivn <int> max bucket sz as divisor of ref len (default: 4)
## --dcv <int> diff-cover period for blockwise (default: 1024)
## --nodc disable diff-cover (algorithm becomes quadratic)
## -r/--noref don't build .3/.4 index files
## -3/--justref just build .3/.4 index files
## -o/--offrate <int> SA is sampled every 2^<int> BWT chars (default: 5)
## -t/--ftabchars <int> # of chars consumed in initial lookup (default: 10)
## --threads <int> # of threads
## --seed <int> seed for random number generator
## -q/--quiet verbose output (for debugging)
## -h/--help print detailed description of tool and its options
## --usage print this usage message
## --version print version information and quit
We need to supply the fasta files of the reference (in this case just one file) and a name for the index. Since this is E coli REL606, we might call it Ec606 for short.
$ bowtie2-build data/genome/ecoli_rel606.fasta data/genome/Ec606
While the index is created, you will see a log of output during the build process.
Align reads to reference genome
The alignment process consists of choosing an appropriate reference genome to map our reads against and then deciding on an aligner. We will use the bowtie2 algorithm asking for four processors (-p 4) and speed over sensitivity.
export BOWTIE2_INDEXES=$(pwd)/data/genome
for file in results/trimmed/*1.trim.fastq.gz ; do
SRR=$(basename $file _1.trim.fastq.gz)
echo running $SRR
bowtie2 -x Ec606 \
--very-fast -p 4\
-1 results/trimmed/${SRR}_1.trim.fastq.gz \
-2 results/trimmed/${SRR}_2.trim.fastq.gz \
-S results/sam/${SRR}.sam
done
Notice that when we run bowtie with paried reads we need to specify the pairs with -1 and -2 arguments.
This will take a while!!
SAM/BAM format
The SAM file, is a tab-delimited text file that contains information for each individual read and its alignment to the genome. We discussed the SAM file format in detail in the lecture; the paper by Heng Li et al. provides a lot more detail on the specification.
The compressed binary version of SAM is called a BAM file. We use this version to reduce size and to allow for indexing, which enables efficient random access of the data contained within the file.
The file begins with a header, which is optional. The header is used to describe source of data, reference sequence, method of alignment, etc., this will change depending on the aligner being used. Following the header is the alignment section. Each line that follows corresponds to alignment information for a single read. Each alignment line has 11 mandatory fields for essential mapping information and a variable number of other fields for aligner specific information. An example entry from a SAM file is displayed below with the different fields highlighted.
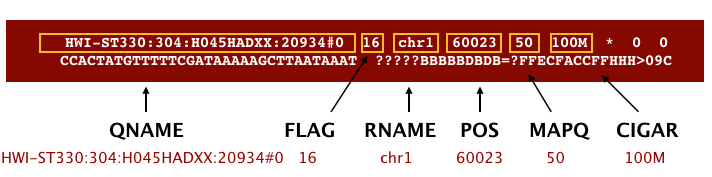
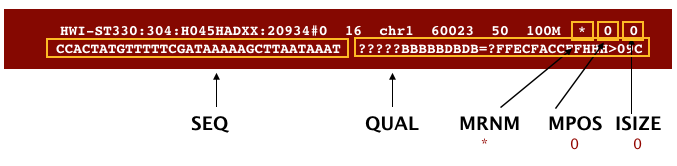
We will convert the SAM file to BAM format using the samtools program with the view command and tell this command that the input is in SAM format (-S) and to output BAM format (-b):
for file in results/sam/*.sam
do
SRR=$(basename $file .sam)
echo $SRR
samtools view -S -b results/sam/${SRR}.sam > results/bam/${SRR}-aligned.bam
done
Sort BAM file by coordinates
Next we sort the BAM file using the sort command from samtools. -o tells the command where to write the output.
for file in results/bam/*-aligned.bam
do
SRR=$(basename $file -aligned.bam)
echo $SRR
samtools sort results/bam/${SRR}-aligned.bam -o results/bam/${SRR}-sorted.bam
done
SAM/BAM files can be sorted in multiple ways, e.g. by location of alignment on the chromosome, by read name, etc. It is important to be aware that different alignment tools will output differently sorted SAM/BAM, and different downstream tools require differently sorted alignment files as input.
You can use samtools to learn more about this bam file as well.
samtools flagstat results/bam/SRR2584866.sorted.bam
Variant calling
A variant call is a conclusion that there is a nucleotide difference vs. some reference at a given position in an individual genome
or transcriptome, often referred to as a Single Nucleotide Polymorphism (SNP). The call is usually accompanied by an estimate of
variant frequency and some measure of confidence. Similar to other steps in this workflow, there are number of tools available for
variant calling. In this workshop we will be using bcftools, but there are a few things we need to do before actually calling the
variants.
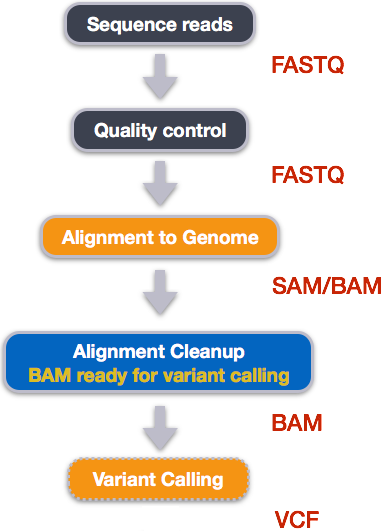
Step 1: Calculate the read coverage of positions in the genome
Do the first pass on variant calling by counting read coverage with bcftools. We will use the command mpileup. The flag -O b tells samtools to generate a bcf format output file, -o specifies where to write the output file, and -f flags the path to the reference genome:
bcftools mpileup -O b -o results/bcf/SRR2584866_raw.bcf \
-f data/genome/ecoli_rel606.fasta results/bam/SRR2584866-sorted.bam
[mpileup] 1 samples in 1 input files
We have now generated a file with coverage information for every base.
Step 2: Detect the single nucleotide polymorphisms (SNPs)
Identify SNPs using bcftools call. We have to specify ploidy with the flag --ploidy, which is
one for the haploid E. coli. -m allows for multiallelic and rare-variant calling, -v tells
the program to output variant sites only (not every site in the genome), and -o specifies where
to write the output file:
bcftools call --ploidy 1 -m -v -o results/bcf/SRR2584866_variants.vcf results/bcf/SRR2584866_raw.bcf
Step 3: Filter and report the SNP variants in variant calling format (VCF)
Filter the SNPs for the final output in VCF format, using vcfutils.pl:
$ vcfutils.pl varFilter results/bcf/SRR2584866_variants.vcf > results/vcf/SRR2584866_final_variants.vcf
Explore the VCF format:
$ less -S results/vcf/SRR2584866_final_variants.vcf
You will see the header (which describes the format), the time and date the file was created, the version of bcftools that was used, the command line parameters used, and some additional information:
##fileformat=VCFv4.2
##FILTER=<ID=PASS,Description="All filters passed">
##bcftoolsVersion=1.8+htslib-1.8
##bcftoolsCommand=mpileup -O b -o results/bcf/SRR2584866_raw.bcf -f data/genome/ecoli_rel606.fasta results/bam/SRR2584866.aligned.sorted.bam
##reference=file://data/ref_genome/ecoli_rel606.fasta
##contig=<ID=CP000819.1,length=4629812>
##ALT=<ID=*,Description="Represents allele(s) other than observed.">
##INFO=<ID=INDEL,Number=0,Type=Flag,Description="Indicates that the variant is an INDEL.">
##INFO=<ID=IDV,Number=1,Type=Integer,Description="Maximum number of reads supporting an indel">
##INFO=<ID=IMF,Number=1,Type=Float,Description="Maximum fraction of reads supporting an indel">
##INFO=<ID=DP,Number=1,Type=Integer,Description="Raw read depth">
##INFO=<ID=VDB,Number=1,Type=Float,Description="Variant Distance Bias for filtering splice-site artefacts in RNA-seq data (bigger is better)",Version=
##INFO=<ID=RPB,Number=1,Type=Float,Description="Mann-Whitney U test of Read Position Bias (bigger is better)">
##INFO=<ID=MQB,Number=1,Type=Float,Description="Mann-Whitney U test of Mapping Quality Bias (bigger is better)">
##INFO=<ID=BQB,Number=1,Type=Float,Description="Mann-Whitney U test of Base Quality Bias (bigger is better)">
##INFO=<ID=MQSB,Number=1,Type=Float,Description="Mann-Whitney U test of Mapping Quality vs Strand Bias (bigger is better)">
##INFO=<ID=SGB,Number=1,Type=Float,Description="Segregation based metric.">
##INFO=<ID=MQ0F,Number=1,Type=Float,Description="Fraction of MQ0 reads (smaller is better)">
##FORMAT=<ID=PL,Number=G,Type=Integer,Description="List of Phred-scaled genotype likelihoods">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##INFO=<ID=ICB,Number=1,Type=Float,Description="Inbreeding Coefficient Binomial test (bigger is better)">
##INFO=<ID=HOB,Number=1,Type=Float,Description="Bias in the number of HOMs number (smaller is better)">
##INFO=<ID=AC,Number=A,Type=Integer,Description="Allele count in genotypes for each ALT allele, in the same order as listed">
##INFO=<ID=AN,Number=1,Type=Integer,Description="Total number of alleles in called genotypes">
##INFO=<ID=DP4,Number=4,Type=Integer,Description="Number of high-quality ref-forward , ref-reverse, alt-forward and alt-reverse bases">
##INFO=<ID=MQ,Number=1,Type=Integer,Description="Average mapping quality">
##bcftools_callVersion=1.8+htslib-1.8
##bcftools_callCommand=call --ploidy 1 -m -v -o results/bcf/SRR2584866_variants.vcf results/bcf/SRR2584866_raw.bcf; Date=Tue Oct 9 18:48:10 2018
Followed by information on each of the variations observed:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT results/bam/SRR2584866.aligned.sorted.bam
CP000819.1 1521 . C T 207 . DP=9;VDB=0.993024;SGB=-0.662043;MQSB=0.974597;MQ0F=0;AC=1;AN=1;DP4=0,0,4,5;MQ=60
CP000819.1 1612 . A G 225 . DP=13;VDB=0.52194;SGB=-0.676189;MQSB=0.950952;MQ0F=0;AC=1;AN=1;DP4=0,0,6,5;MQ=60
CP000819.1 9092 . A G 225 . DP=14;VDB=0.717543;SGB=-0.670168;MQSB=0.916482;MQ0F=0;AC=1;AN=1;DP4=0,0,7,3;MQ=60
CP000819.1 9972 . T G 214 . DP=10;VDB=0.022095;SGB=-0.670168;MQSB=1;MQ0F=0;AC=1;AN=1;DP4=0,0,2,8;MQ=60 GT:PL
CP000819.1 10563 . G A 225 . DP=11;VDB=0.958658;SGB=-0.670168;MQSB=0.952347;MQ0F=0;AC=1;AN=1;DP4=0,0,5,5;MQ=60
CP000819.1 22257 . C T 127 . DP=5;VDB=0.0765947;SGB=-0.590765;MQSB=1;MQ0F=0;AC=1;AN=1;DP4=0,0,2,3;MQ=60 GT:PL
CP000819.1 38971 . A G 225 . DP=14;VDB=0.872139;SGB=-0.680642;MQSB=1;MQ0F=0;AC=1;AN=1;DP4=0,0,4,8;MQ=60 GT:PL
CP000819.1 42306 . A G 225 . DP=15;VDB=0.969686;SGB=-0.686358;MQSB=1;MQ0F=0;AC=1;AN=1;DP4=0,0,5,9;MQ=60 GT:PL
CP000819.1 45277 . A G 225 . DP=15;VDB=0.470998;SGB=-0.680642;MQSB=0.95494;MQ0F=0;AC=1;AN=1;DP4=0,0,7,5;MQ=60
CP000819.1 56613 . C G 183 . DP=12;VDB=0.879703;SGB=-0.676189;MQSB=1;MQ0F=0;AC=1;AN=1;DP4=0,0,8,3;MQ=60 GT:PL
CP000819.1 62118 . A G 225 . DP=19;VDB=0.414981;SGB=-0.691153;MQSB=0.906029;MQ0F=0;AC=1;AN=1;DP4=0,0,8,10;MQ=59
CP000819.1 64042 . G A 225 . DP=18;VDB=0.451328;SGB=-0.689466;MQSB=1;MQ0F=0;AC=1;AN=1;DP4=0,0,7,9;MQ=60 GT:PL
This is a lot of information, so let’s take some time to make sure we understand our output.
The first few columns represent the information we have about a predicted variation.
| column | info |
|---|---|
| CHROM | contig location where the variation occurs |
| POS | position within the contig where the variation occurs |
| ID | a . until we add annotation information |
| REF | reference genotype (forward strand) |
| ALT | sample genotype (forward strand) |
| QUAL | Phred-scaled probablity that the observed variant exists at this site (higher is better) |
| FILTER | a . if no quality filters have been applied, PASS if a filter is passed, or the name of the filters this variant failed |
In an ideal world, the information in the QUAL column would be all we needed to filter out bad variant calls.
However, in reality we need to filter on multiple other metrics.
The last two columns contain the genotypes and can be tricky to decode.
| column | info |
|---|---|
| FORMAT | lists in order the metrics presented in the final column |
| results | lists the values associated with those metrics in order |
For our file, the metrics presented are GT:PL:GQ.
| metric | definition |
|---|---|
| GT | the genotype of this sample which for a diploid genome is encoded with a 0 for the REF allele, 1 for the first ALT allele, 2 for the second and so on. So 0/0 means homozygous reference, 0/1 is heterozygous, and 1/1 is homozygous for the alternate allele. For a diploid organism, the GT field indicates the two alleles carried by the sample, encoded by a 0 for the REF allele, 1 for the first ALT allele, 2 for the second ALT allele, etc. |
| PL | the likelihoods of the given genotypes |
| GQ | the Phred-scaled confidence for the genotype |
| AD, DP | the depth per allele by sample and coverage |
The Broad Institute’s VCF guide is an excellent place to learn more about VCF file format.
Exercise
Use the
grepandwccommands you’ve learned to assess how many variants are in the vcf file.Solution
$ grep -v "#" results/vcf/SRR2584866_final_variants.vcf | wc -l804There are 804 variants in this file.
Assess the alignment (visualization) - optional step
It is often instructive to look at your data in a genome browser. Visualisation will allow you to get a “feel” for the data, as well as detecting abnormalities and problems. Also, exploring the data in such a way may give you ideas for further analyses. As such, visualization tools are useful for exploratory analysis. In this lesson we will describe two different tools for visualisation; a light-weight command-line based one and the Broad Institute’s Integrative Genomics Viewer (IGV) which requires software installation and transfer of files.
In order for us to visualize the alignment files, we will need to index the BAM file using samtools:
$ samtools index results/bam/SRR2584866-sorted.bam
Viewing with tview
Samtools implements a very simple text alignment viewer based on the GNU
ncurses library, called tview. This alignment viewer works with short indels and shows MAQ consensus.
It uses different colors to display mapping quality or base quality, subjected to users’ choice. Samtools viewer is known to work with an 130 GB alignment swiftly. Due to its text interface, displaying alignments over network is also very fast.
In order to visualize our mapped reads we use tview, giving it the sorted bam file and the reference file:
$ samtools tview results/bam/SRR2584866-sorted.bam data/genome/ecoli_rel606.fasta
1 11 21 31 41 51 61 71 81 91 101 111 121
AGCTTTTCATTCTGACTGCAACGGGCAATATGTCTCTGTGTGGATTAAAAAAAGAGTGTCTGATAGCAGCTTCTGAACTGGTTACCTGCCGTGAGTAAATTAAAATTTTATTGACTTAGGTCACTAAATAC
..................................................................................................................................
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, ..................N................. ,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,........................
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, ..................N................. ,,,,,,,,,,,,,,,,,,,,,,,,,,,.............................
...................................,g,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, .................................... ................
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,.................................... .................................... ,,,,,,,,,,
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, .................................... ,,a,,,,,,,,,,,,,,,,,,,,,,,,,,,,, .......
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, ............................. ,,,,,,,,,,,,,,,,,g,,,,, ,,,,,,,,,,,,,,,,,,,,,,,,,,,,
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, ...........................T....... ,,,,,,,,,,,,,,,,,,,,,,,c, ......
......................... ................................ ,g,,,,,,,,,,,,,,,,,,, ...........................
,,,,,,,,,,,,,,,,,,,,, ,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, ,,,,,,,,,,,,,,,,,,,,,,,,,,, ..........................
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, ................................T.. .............................. ,,,,,,
........................... ,,,,,,g,,,,,,,,,,,,,,,,, .................................... ,,,,,,
,,,,,,,,,,,,,,,,,,,,,,,,,, .................................... ................................... ....
.................................... ........................ ,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, ....
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, ,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, ,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,
........................ .................................. ............................. ....
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, .................................... ..........................
............................... ,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, ....................................
................................... ,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, ,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, ,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, ..................................
.................................... ,,,,,,,,,,,,,,,,,,a,,,,,,,,,,,,,,,,, ,,,,,,,,,,,,,,,,,,,,,,,,,
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,, ............................ ,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,
The first line of output shows the genome coordinates in our reference genome. The second line shows the reference
genome sequence. The third lines shows the consensus sequence determined from the sequence reads. A . indicates
a match to the reference sequence, so we can see that the consensus from our sample matches the reference in most
locations. That is good! If that wasn’t the case, we should probably reconsider our choice of reference.
Below the horizontal line, we can see all of the reads in our sample aligned with the reference genome. Only
positions where the called base differs from the reference are shown. You can use the arrow keys on your keyboard
to scroll or type ? for a help menu. To navigate to a specific position, type g. A dialogue box will appear. In
this box, type the name of the “chromosome” followed by a colon and the position of the variant you would like to view
(e.g. for this sample, type CP000819.1:50 to view the 50th base. Type Ctrl^C or q to exit tview.
Exercise
Visualize the alignment of the reads for our
SRR2584866sample. What variant is present at position 4377265? What is the canonical nucleotide in that position?Solution
$ samtools tview ~/dc_workshop/results/bam/SRR2584866.aligned.sorted.bam ~/dc_workshop/data/ref_genome/ecoli_rel606.fastaThen type
g. In the dialogue box, typeCP000819.1:4377265.Gis the variant.Ais canonical. This variant possibly changes the phenotype of this sample to hypermutable. It occurs in the gene mutL, which controls DNA mismatch repair.
Viewing with IGV
IGV is a stand-alone browser, which has the advantage of being installed locally and providing fast access. Web-based genome browsers, like Ensembl or the UCSC browser, are slower, but provide more functionality. They not only allow for more polished and flexible visualisation, but also provide easy access to a wealth of annotations and external data sources. This makes it straightforward to relate your data with information about repeat regions, known genes, epigenetic features or areas of cross-species conservation, to name just a few.
In order to use IGV, we will need to transfer some files to our local machine. We know how to do this with scp.
Open a new tab in your terminal window and create a new folder. We’ll put this folder on our Desktop for
demonstration purposes, but in general you should avoide proliferating folders and files on your Desktop and
instead organize files within a directory structure like we’ve been using in our dc_workshop directory.
$ mkdir ~/Desktop/files_for_igv
$ cd ~/Desktop/files_for_igv
Next we need to open the IGV software. If you haven’t done so already, you can download IGV from the Broad Institute’s software page, double-click the .zip file
to unzip it, and then drag the program into your Applications folder.
- Open IGV.
- Load our reference genome file (
ecoli_rel606.fasta) into IGV using the “Load Genomes from File…“ option under the “Genomes” pull-down menu. - Load our BAM file (
SRR2584866-sorted.bam) using the “Load from File…“ option under the “File” pull-down menu. - Do the same with our VCF file (
SRR2584866_final_variants.vcf).
Your IGV browser should look similar to the screenshot below:
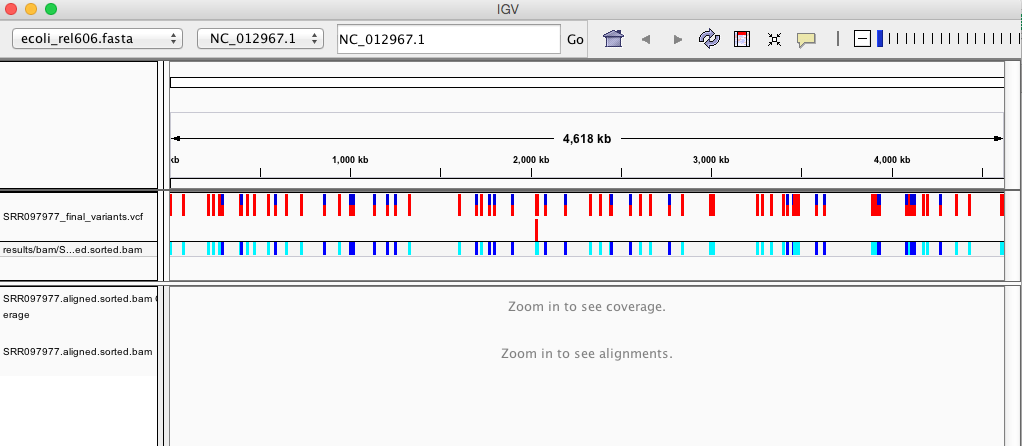
There should be two tracks: one coresponding to our BAM file and the other for our VCF file.
In the VCF track, each bar across the top of the plot shows the allele fraction for a single locus. The second bar shows the genotypes for each locus in each sample. We only have one sample called here so we only see a single line. Dark blue = heterozygous, Cyan = homozygous variant, Grey = reference. Filtered entries are transparent.
Zoom in to inspect variants you see in your filtered VCF file to become more familiar with IGV. See how quality information corresponds to alignment information at those loci. Use this website and the links therein to understand how IGV colors the alignments.
Now that we’ve run through our workflow for a single sample, we want to repeat this workflow for our other five samples. However, we don’t want to type each of these individual steps again five more times. That would be very time consuming and error-prone, and would become impossible as we gathered more and more samples. Luckily, we already know the tools we need to use to automate this workflow and run it on as many files as we want using a single line of code. Those tools are: wildcards, for loops, and bash scripts. We’ll use all three in the next lesson.
Other Notes
Multi-line commands
Some of the commands we ran in this lesson are burly! When typing a long
command into your terminal, you can use the \ character
to separate code chunks onto separate lines. This can make your code more readable.
Key Points
Bioinformatics command line tools are collections of commands that can be used to carry out bioinformatics analyses.
To use most powerful bioinformatics tools, you’ll need to use the command line.
There are many different file formats for storing genomics data. It’s important to understand what type of information is contained in each file, and how it was derived.